- Documentation Main Page
- Creation of the Cancer Gene Index
- Data, Metadata, and Annotations
- Cancer Gene Index Gene-Disease and Gene-Compound XML Documents
- caBIO APIs
- Cancer Gene Index Shared Parsed Data and Code
- caBIO Portlet Templated Searches
- caBIO Home Page
- caBIO iPhone Application
- caBIO Portlet Simple Searches
- Glossary
- Credits and Resources
To Print the Guide
We recommend you print one wiki page of the guide at a time. To do this, click the printer icon at the top right of the page; then from the browser File menu, choose Print. Printing multiple pages at one time is more complex. For instructions, refer to Printing multiple pages .
Having Trouble Reading the Text?
Resizing the text for any web page is easy. For information on how to do this in your web browser, refer to this W3C tutorial 
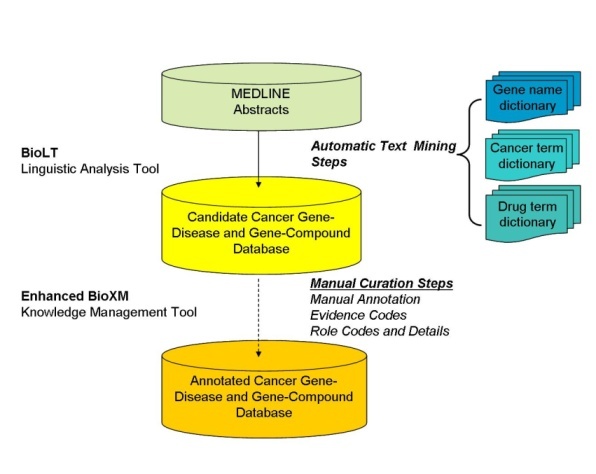
Creation of the Cancer Gene Index Overview
As part of the caBIG® initiative, the NCI contractor Sophic Systems Alliance, Inc.
and their strategic partner Biomax™ Informatics AG
created the Cancer Gene Index by using mining millions of MEDLINE abstracts with a combination of automated linguistic text analysis and manual validation and annotation by expert curators.
This large-scale project was an iterative effort that was completed over six phases. Although the artifacts (for example, text files or XML) from each phase are available on the Cancer Gene Index web page, the sixth phase represents the final Cancer Gene Index data source.
Lexical Dictionaries
Despite significant effort to standardize nomenclature for biological entities, multiple terminologies for the same compound, disease, or gene are often found within the scientific literature. These terms may include names, acronyms, abbreviations, or spelling variations that are outdated, either because of the age of a publication or because, even in newer publications, scientists may continue to use older terms with which they are familiar. In addition, researchers may invent new terms for biological concepts (for example, the actual compound, disease, or gene to which the various names, acronyms, and abbreviations refer) with established names, and this phenomenon is seen frequently for genes. Thus, often many different aliases refer to the same biological concept. This diverse usage of terms makes it difficult to extract data from abstracts using any one reference concept name (for example, Entrez or Ensembl gene name).
In order to mine the MEDLINE abstracts for sentences that contained information about gene-disease or gene-compound associations, lexical dictionaries were created from the NCI Thesaurus, public name catalogs, public classifications, and terms from the MEDLINE abstracts. The Compound Term Dictionary includes the NCI Thesaurus Pharmacologic Substance concept and all of its child sub-concepts, Pharmacologic Substance synonym terminologies and their sub-concepts, and any concept terminology that had the NCI Thesaurus semantic type property "Pharmacologic Substance." The Cancer Term Dictionary was created from public disease term catalogs, public disease classifications, and terms used within the literature. For the last case, terms within the publications were extracted by spelling variation identification, acronym recognition, and disambiguation procedures. Disease terms from the three sources were mapped to disease terminologies in the NCI Thesaurus and combined in a non-redundant fashion. The Cancer Term Dictionary had approximately 80,000 unique cancer disease term entries covering all of the disease terminologies from the various sources. The Gene Term Dictionary was based the union of HUGO Gene Nomenclature Committee (HGNC), LocusLink, and the Gene Database (GDB) data. These data were augmented with gene terms from the literature using sophisticated, automated procedures that performed spelling variation identification, acronym recognition, disambiguation, and context-based gene name recognition as described in the Sophic and Biomax™ Cancer Gene Index white paper white paper
This union resulted in a total of 350,000 unique gene name entries from the three reference sources, which resolved to less than 10,000 unique genes.
Each dictionary term is linked back to a unique caBIG® Enterprise Vocabulary Services (EVS) concept code.
MEDLINE Abstract Text Mining
The Biomax™ LT Linguistic Analysis Tool, a natural language processing tool, was used to mine the text of approximately 90 million total sentences from nearly 20 million MEDLINE abstracts to select those sentences that contained meaningful information about associations between gene names and disease or compound terms (for example, contained terms from both the Gene and Disease Term or the Gene and Compound Term lexical dictionaries). Biomax™ LT is a sensitive text mining tool, such that any sentence with a putative gene-disease or gene-compound relationship was not omitted during the automated text mining phase. These candidate sentences were stored in an enhanced version of the BioXM Knowledge Management database, so that human curators could later manually validate and annotate them with Evidence Codes, Role Codes, Role Details, and other information.
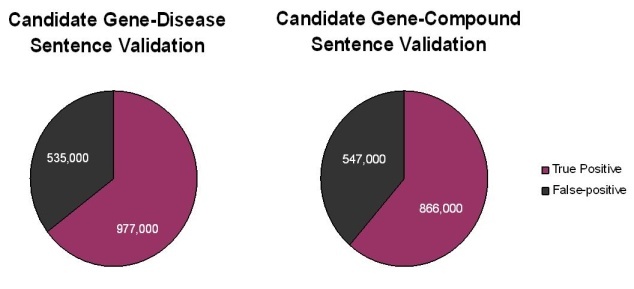
A total of 1.5 million putative gene-disease and 1.4 million putative gene-compound sentences and their PubMed Reference Identifiers were extracted by Biomax LT. Subsequent careful reading of these sentences by expert human curators showed that approximately two-thirds of the sentences extracted by the Biomax LT tool were correct. The remaining sentences were false positives that occurred not because the automated algorithm misidentified a term from the dictionaries, but rather because of context. Many of the "false" positives resulted from ambiguous acronyms (for example, HCC can be FAM 126A gene synonym or hepatocellular carcinoma disease name) or from gene names being synonymous for multiple gene concept codes (for example, p63 is a valid synonym for the three concepts TP63, CKAP4, and UVRAG).
Knowledge Management
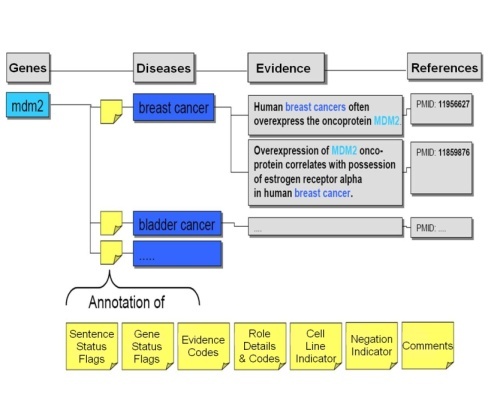
As the candidate gene-disease and gene-compound sentences were extracted by Biomax™ LT, they were stored in a relational database pending validation and annotation. The BioXM Annotation Module, part of the Biomax™ integrated knowledge management suite, provided the distributed annotation environment infrastructure for these steps. BioXM was extended in order to accommodate the massive volume of extracted data from Biomax™ LT, the large number of curators, and the continuous input of their annotations. The BioXM Module also facilitated the use of controlled vocabularies for these annotations.
Ph.D.-level curators carefully read each sentence to validate that the sentence truly contained evidence of gene-disease or gene-compound associations. The curators also annotated sentences with descriptions of the nature of the gene-compound or gene-disease relationship and of the evidence in the sentence from which the relationship was determined. In addition, the curators set flags for genes (#Gene Status Flags) and sentences (#Sentence Status Flags) to describe their status, whether or not the evidence was from a cell line or was a negative finding (i.e, gene X is NOT associated with disease Y), and also often gave free-text #comments on records. This process is outlined in the following figure, which was adapted from the Biomax™ Informatics, AG Cancer Gene Index white paper
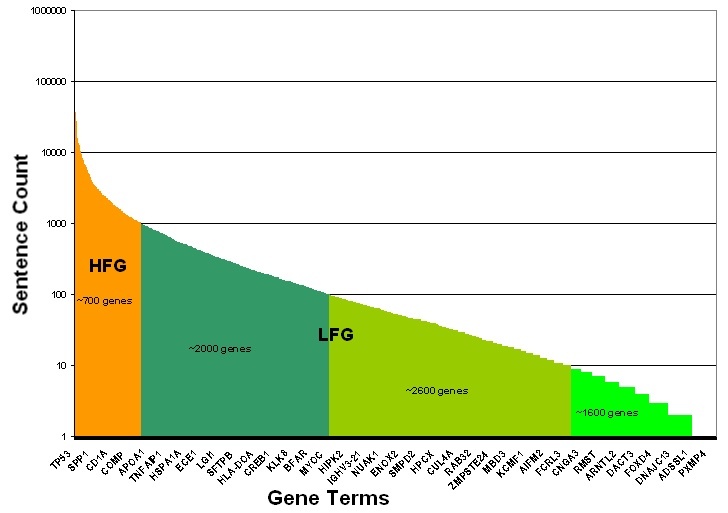
High-Frequency Genes
Cancer genes were classified as low- or high-frequency genes based upon the total number of extracted sentences in which they were mentioned, as illustrated in the following figure Adapted from the Cancer Gene Index Project poster. Well-studied cancer genes with greater than 1,000 MEDLINE abstract sentences containing gene-disease or gene-compound associations were categorized as High Frequency Sentence Count Genes (HFG, orange). Cancer genes with fewer than 1,000 sentences were categorized as Low Frequency Sentence Count Genes (LFG, shades of green). LFGs are binned into three categories based upon the number of sentences in which they were associated with a disease or compound term: 1-10 (lime green), 11-100 (avocado green), or 101-1,000 (forest green) sentence counts. Exemplar HFG and LFG genes are shown on the x-axis, as there are too many gene terms to display all term names. Whereas the LFGs were manually validated and curated, the large number of sentences for HFGs required the development of a filtering approach to determine which of the HFG sentences would be manually annotated. The approach ensured that all associations were captured without the need to recursively curate well-documented associations.
All low-frequency genes were validated and annotated by expert curators as described above. Because in some cases, high-frequency genes had thousands of associated sentences, manual curation for all of these sentences was not feasible. A rational filtering approach, shown in the flowchart below, was therefore applied to the sentences describing high frequency gene relationships. The sentences containing information about a high frequency gene were categorized as having gene-disease (GD, blue shapes), gene-compound (GC, gray shapes) relationship, or in some cases, both (green shapes). Analysis showed that ~80% of the sentences describing gene-disease or gene-compound associations could be fully manually annotated. The remaining ~20% of sentences could not easily be fully manually annotated, because there were too many associated sentences to complete the manual steps in a reasonable time frame, as illustrated in the following two figures.
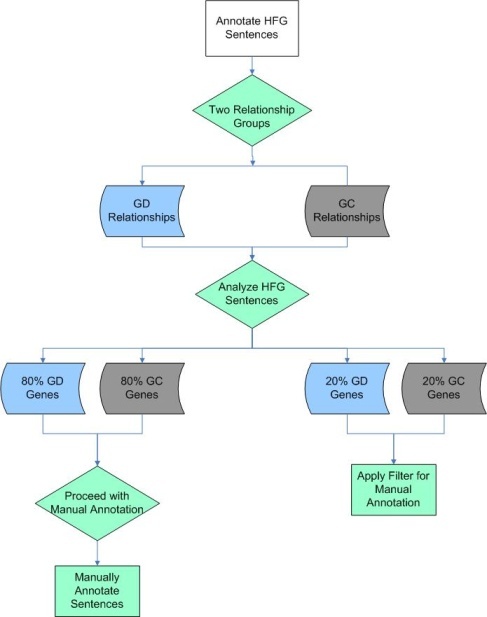
High Frequency Gene Filtering
The development of an annotation filtering methodology began with a natural language processing analysis of candidate high frequency gene-disease sentences, with gene-compound sentence analysis being postponed until gene-disease sentence filtering and annotation was complete. The analysis showed that 99% of the gene-disease sentences described Expression-Gene Relationship, Abnormality-Gene Relationship (B), Biomarker-Gene Relationship, or Therapy-Gene Relationship. Gene-disease sentences were then categorized by "quadrants" based on whether all four relationships were described within the sentence (Q1), three were described (Q2), two (Q3) or only one (Q4). All Q1 and Q2 sentences, in which three or four of the gene-disease relationships co-occurred, were manually annotated. Additional filtering criteria were applied to Q3 and Q4 sentences, which described only one or two of the relationship categories. The criteria for this second filtering step were to only include sentences from the three or four most recent articles that were published in high impact journals and that had a high citation index. The Q3 and Q4 sentences that matched these filtering criteria were flagged for manual curation.
Once filtering of gene-disease sentences was complete, a similar procedure was followed for sentences with gene-compound associations. Natural language processing analysis of these candidate sentences showed that the vast majority of these associations could be classified as describing Binding (A*), Regulation (B*), and Resistance (C*). Sentences where binding, regulation, and resistance co-occur were all manually annotated. Sentences with an occurrence of one or two of the categories were filtered with the impact factor and publication date criteria, as before.
Please refer to the High Frequency Gene Filtering Workflow page to view the filtering flowchart.
Generation of Cancer Gene Index Gene-Disease and Gene-Compound XML
Sentence validation information and annotations were added by the human curators to the BioXM database, and flags were set by the curators to indicate where in the this process each sentence and gene fell. Sentence flags indicated that the sentence had been reviewed and whether annotation was complete. Gene flags indicated whether annotations were complete for all gene-disease or gene-compound sentences that include the gene concept (for example, inclusive of all synonyms and nomenclature variations). The final Cancer Gene Index Gene-Disease and Gene-Compound XML files were created from this database.
Attribution
The NCI established caBIG® to accelerate the discovery of efficacious methods for cancer detection, diagnostics, treatment, and prevention in order to ultimately improve patient outcomes. caBIG® is a network that links researchers, physicians, and patients throughout the cancer community and that provides standard data elements, rules, terminologies, and vocabularies to facilitate the sharing of data and information through interoperable infrastructure. These terminology and vocabulary standards are implemented in the Cancer Gene Index, as well as in a variety of interoperable, reference life science and clinical research data management and analysis software applications. The Cancer Gene Index was created by the contractors Biomax™ Informatics AG and their partner Sophic Systems Alliance, Inc. with additional project management and oversight by the NCI and SAIC-Frederick. A complete attribution is available in Credits and Resources.