Question: The datafile is imported, but it is not downloadable. Why?
Topic: caArray Installation and Upgrade
Release: caArray 2.4.0
Date entered: 04/20/2011
Details about the Question
We had previously installed caArray - v2.2.1, and UPT - v3.2.0. Recently, we added caIntegrator2 - v1.0 on Redhat 5. They are all on one machine using the same mysql instance. However, we couldn't provision additional caIntegrator2 users.
We have uploaded CHP and CEL files (as well as supplemental files) for 72 patients into our instance of caArray - this process was done manually, not using MAGE-TAB. All 144 CHP/CEL files have been successfully imported and validated (green check mark). When I watched the tutorial video for uploading caArray data, I noticed that once the files had been imported and validated, they no longer showed up under the "Manage Data" tab but are all under "Imported Data" or "Supplemental Files." However, in our case, the files are also under "Manage Data" in addition to "Imported Data." Was our data somehow not imported correctly?
Additionally, when I go to the "Annotations"/"Samples" tab, I can only download files for 3 of the samples - the rest of them display a message when clicked: "There was no data associated with the sample you requested".
Answer
The import process is the last step of data uploading in caArray, which allows the array data to be stored in the database. During the importing process, caArray associates the data with the appropriate biomaterial and hybridization annotation by creating an annotation chain of source TO sample TO extract TO labeled extract TO hybridization. The array data can be downloaded only if the data were associated properly.
Import MAGE-TAB set
If a MAGE-TAB set (IDF and SDRF) is imported along with the data files, where the SDRF file refers to each of the data files, caArray will use the information provided in SDRF to determine how to create sources, samples, extracts, labeled extracts and hybridizations.
Import Only the datafile
If only data files (for example, .cel, .chp, etc.) are imported, caArray offers three options to associate the data and annotation.
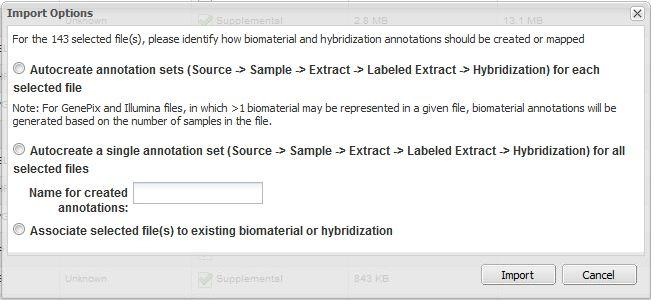
Option 1. Autocreate annotation sets ... for each selected file
For every unique file name to be imported, caArray automatically creates a Source - Sample - Extract - Labeled Extract - Hybridization chain corresponding to each data file imported.
Option 2. Autocreate a single annotation set ... for all selected files
caArray creates a single Source - Sample - Extract - Labeled Extract - Hybridization chain, and associates all selected data files with this single chain.
Option 3. Associate selected file(s) to existing biomaterial or hybridization
caArray displays all available sources, samples, extracts, labeled extracts and hybridizations. The user selects one of these, and caArray associates the selected files with that biomaterial or hybridization. Note that additional items in the chain (to the right of the selected biomaterial) may need to be generated by the system.
Troubleshooting: Choose the right option
The proper association between the array data and annotation data is critical to a successful array data import.
The annotation data might be created before the data files were uploaded and imported. These annotation data, however, have to be used properly. For example, when the new biomaterial objects are created during the auto-create process, the biomaterial data manually entered would be ignored and associated with no datafiles.
If there are annotation data manually entered to the experiment prior to the importing process, the user should choose option 3 to select the hybridization object during the import so that there would be no downstream creation of biomaterial objects.
If the user chooses one of the options of "Autocreate...", there should be no manual biomaterial annotation entered.
Have a comment?
Please leave your comment in the caArray End User Forum.